Si tienes Amelogénesis Imperfecta, ¿Por qué deberías comprar ENAMELIN?
La Amelogénesis Imperfecta se transmite por genética. Se presenta con extremamente mala formación del esmalte. El mal funcionamiento de las proteínas en el esmalte (Ameloblastina, Enamelina, Tuftelina y Amelogenina) es la causa de Amelogénesis Imperfecta. La mayoría de los casos se presenta con dientes hipersensibles e Hipoplasia Del Esmalte.
Deberías comprar ENAMELIN porque ENAMELIN remineraliza los dientes, fortalece los dientes y elimina la hipersensibilidad por completo. ENAMELIN es la mejor solución para personas con dientes hipersensibles e Hipoplasia Del Esmalte. ENAMELIN es “La mejor solución para personas con Amelogénesis Imperfecta”.
Más información sobre Amelogénesis Imperfecta
Amelogénesis imperfecta
La amelogénesis imperfecta (AI) es una enfermedad genética que afecta la estructura y la apariencia del esmalte de los dientes.[1] Los dientes son muy pequeños, decolorados, quebradizos o apiñados, y propensos a un rápido desgaste con caries y pérdida temprana de los dientes. Estos problemas dentales, que varían entre las personas afectadas, pueden afectar tanto a los dientes primarios (de los bebés) como a los dientes permanentes. Las personas con esta enfermedad también tienen problemas relacionados con los tejidos que rodean los dientes (tejidos periodontales) como las encías, el cemento, los ligamentos, y los huesos alveolares en los que descansa la raíz del diente. Los dientes también son sensibles a las temperaturas calientes o frías, y a veces a ambas. En algunos casos hay un dolor severo y continuo debido a la dentina expuesta que resulta del defecto del esmalte.[2][3]
Hay 4 tipos principales de AI que se clasifican según los síntomas, la apariencia de los rayos X y el tipo de defecto del esmalte. Los principales tipos son: hipoplásico (tipo I); tipo con hipomaturación (tipo II); hipocalcificado (tipo III); e hipomaturación / hipoplasia / taurodontismo (tipo IV). Estos 4 tipos se dividen en 17 o 18 subtipos, que se distinguen por el tipo de variantes patogénicos (mutaciones) específicas que causan la enfermedad y por su patrón de herencia.[4][3] Las variantes que causan la enfermedad resultan en ameloblastos (células encargadas de la formación y organización del esmalte dental) anormales que producen un esmalte anormal o que no producen esmalte.[5][6] La herencia de la AI puede ser autosómica dominante, autosómica recesiva, o recesiva ligada al cromosoma X.[2] El tratamiento puede incluir dentaduras que sellan los dientes con defectos (restauraciones completas de la corona del diente), tratamiento de ortodoncia, pasta de dientes especial para la sensibilidad de los dientes, y una buena higiene oral.[3][7]
Última actualización: 6/11/2018
¿Usted tiene información actualizada sobre esta condición? ¡Nos gustaría recibir sus comentarios!
Síntomas Síntomas
De forma general, el esmalte de los dientes es blando y delgado. Los dientes se ven amarillos y se dañan fácilmente, y tanto los dientes de leche (dentición primaria) como los dientes permanentes pueden ser afectados.[1]
Los defectos en amelogénesis imperfecta son bastante variables e incluyen anomalías clasificadas en: hipoplásticas (defectos en la cantidad de esmalte), hipomaturación (defecto en el crecimiento final y maturación del esmalte), e hipocalcificación (defecto en la formación del esmalte inicial seguidos de defecto de crecimiento). El esmalte en los tipos de hipomaturación e hipocalcificación no está mineralizado y se describe como hipomineralizado.[5]
El diagnóstico y clasificación de amelogénesis imperfecta tradicionalmente se basa en la presentación clínica y en la forma de herencia. Hay 4 tipos principales basados en los defectos del esmalte y en las señales y síntomas. Estos tipos se subdividen en por lo menos 17-18 diferentes subtipos basados en la variante causante de la enfermedad y en el modo de herencia.[5][3]
-
Tipo I - Hipoplástico: Los dientes son pequeños, pueden estar mal implantados y el color puede ser normal, blanco opaco o café-amarillento. El esmalte varia, pudiendo ser muy delgado y liso o con espesura normal pero con hendiduras, surcos o agujeros. La herencia es autosómica dominante, autosómica recesiva, o ligado al X.
-
Tipo II - Hipomaturación: El aspecto de los dientes es variado y pueden tener una coloración crema-opaca o un color marcadamente café/amarillo, la superficie del diente puede ser suave o rugosa. La sensibilidad de los dientes esta aumentada y la mala oculsión. El esmalte tiene espesura normal pero se quiebra y daña fácilmente. La herencia es autosómica dominante, autosómica recesiva, o ligada al X.
-
Tipo III - Hipocalcificado: El color de los dientes varía entre un blanco-opaco a un amarillo-café. La superficie del esmalte es suave o áspera, la sensibilidad de los dientes esta aumentada, la malaoclusión es común. Hay formación de placa. El esmalte tiene espesura normal pero se quiebra y daña fácilmente. La herencia es autosómica dominante o autosómica recesiva.
-
Tipo IV - Hipomaturación/Hipoplasia/Taurodontismo: El color puede ser blanco-amarillado con puntos café. Los dientes son pequeños y mal implantados. El esmalte tiene espesura reducida, es hipomineralizado en algunas áreas y tiene agujeros. La herencia es autosómica dominante.
Las señales y los síntomas asociados con la amelogénesis imperfecta son similares a los problemas dentales que a veces son parte de algunos desordenes como nefrocalcinosis, linfagiectasia intestinal, citomegalovirus, síndrome alcohólica fetal, uso de tetraciclina, uso de nicotina, leucodermia, y prematuridad.[5]
Última actualización: 6/11/2018
Los diferentes tipos de amelogenesis imperfecta son causados por variantes patogénicos (mutaciones) en diferentes genes:[3]
-
El tipo IA es causado por mutaciones en el gen LAMB3
-
El tipoIB es causado por mutaciones en el gen ENAM
-
El tipo IC es causado por mutaciones en el gen ENAM
-
El tipo 1E es causado por mutaciones en el gen AMELX.
-
El tipo IF es causado por mutaciones en el gen AMBN
-
El tipo IG o síndrome esmalte-renales es causado por mutaciones en el gen FAM20A
-
El tipo IH es causado por mutaciones en el gen ITGB6
-
El tipo IJ es causado por mutaciones en el gen ACPT
-
El tipo IIA1 es causado por mutaciones en el gen KLK4
-
El tipo IIA2 es causado por mutaciones en el gen MMP20
-
El tipo IIA3 es causado por mutaciones en el gen WDR72
-
El tipo IIA4 es causado por mutaciones en el gen ODAPH
-
El tipo IIA5 es causado por mutaciones en el gen SLC24A4
-
El tipo IIA6 es causado por mutaciones en el gen GPR68
-
El tipo IIIA es causado por mutaciones en el gen FAM83H
-
El tipo IIIB es causado por mutaciones en el gen AMTN
-
El tipo IV es causado por mutaciones en el gen DLX3
En algunos casos no se ha identificado el gen mutado por lo que hay investigaciones que tratan de identificar otros genes que pueden estar alterados. Muchos de estos genes proporcionan instrucciones para hacer proteínas responsables por la formación del esmalte. Una mutación en estos genes resulta en ausencia o cambio de la estructura de estas proteínas. Cuando no hay proteínas o cuando no funcionan bien el esmalte no es normal sino que tiene espesura reducida o no es duro, o tiene color amarillo o café, está debilitado, y es fácilmente dañado.[2]
Última actualización: 6/12/2018
La amelogénesis imperfecta puede tener diferentes tipos de herencia de acuerdo al gen que esta alterado:[3]
Los siguientes tipos tienen herencia autosómica dominante:
-
El tipo IA
-
El tipo IB
-
El tipo IIIA
-
El tipo IIIB
-
El tipo IV
Los tipos que se heredan de forma autosómica recesiva son:
-
El tipo IC
-
El tipo IF
-
El tipo IG o síndrome esmalte-renal
-
El tipo IH
-
El tipo IJ
-
El tipo IIA1
-
El tipo IIA2
-
El tipo IIA3
-
El tipo IIA4
-
El tipo IIA5
-
El tipo IIA6
El tipo que se hereda en un patrón dominante ligado al cromosoma X:
-
El tipo IE
Los seres humanos tienen 23 pares de cromosomas y en cada cromosoma hay muchos genes, que tienen la información genética. Los genes, como los cromosomas, vienen en pares, un gen en cada par viene de la madre y el otro del padre. Un trastorno autosómico recesivo significa que para ser afectado se debe tener dos copias de un gen anormal. Tanto el padre como la madre tienen una copia del gen anormal (son portadores) pero no son afectados por el trastorno y normalmente no tienen señales o síntomas. La chance de que tener un hijo o hija afectado(a) con un trastorno recesivo es de 25% para cada embarazo.
En una enfermedad autosómica dominante, si se hereda una copia del gen anormal la persona puede ser afectada. En general, la copia anormal es heredada de uno de los padres, que también tiene la enfermedad. Una persona afectada con una enfermedad autosómica dominante tiene 50% de chances de tener un hijo o hija afectado(a) en cada embarazo.
Se dice que los genes en el cromosoma X están ligados a X. Los genes ligados a X tienen patrones de herencia distintos porque están presentes en diferentes números en las mujeres (XX) y en los varones (XY). Los trastornos genéticos humanos ligados a X son mucho más comunes en los varones que en las mujeres debido al patrón de herencia ligado a X.
Otros casos resultan de mutaciones nuevas (de novo) en estos genes y ocurren en personas donde no hay otros afectados por este problema en la familia. En algunos casos no se identifica mutaciones en ninguno de estos genes.[2]
Última actualización: 6/12/2018
Un dentista puede identificar y diagnosticar amelogénesis imperfecta con base en la información de la historia de la familia del paciente y con la observación de las señales y síntomas del individuo afectado.[8][2]
Radiografías extra-orales de los dientes pueden revelar la presencia de dientes que no salieron o que fueron absorbidos. Radiografías intra-orales muestran contraste entre el esmalte y la dentina en casos en que la mineralización ha sido afectada.[8]
Vea la lista de testes que hay y de los laboratorios que ofrecen el examen de genética para la amelogénesis imperfecta ofrecida por Genetic Testing Registry. La mayoría de las veces los laboratorios no aceptan contacto directo con los pacientes y sus familias solamente con un profesional de la salud. El profesional de genética puede orientar para saber si se necesita hacer el examen genético.
American Academy of Pediatric Dentistry es una buena fuente de información para encontrar un dentista pediátrico. Para localizar un dentista pediátrico en su área usted puede contactar la siguiente asociación: National Dental Association.
Última actualización: 6/12/2018
El tratamiento de los diferentes tipos de amelogénesis imperfecta depende de la edad de la persona afectad, y de la condición del esmalte afectado. Los tratamientos incluyen el cuidado preventivo usando sellantes y restauración adhesiva por motivos estéticos, reconstrucción prostética con aparatos fijos o removibles y cirugías. Por ejemplo, restauración adhesiva es más efectiva para restaurar los dientes de los tipos de amelogénesis imperfecta hipoplástica. Las formas con hipomineralización y esmalte débil (hipocalcificación, hipomaturación) son muy propensas a fracturas y sensibles al calor y químicos y requieren un tratamiento precoz con coronas dentales completas para que el paciente pueda masticar y realizar sus procedimientos higiénicos adecuadamente.[5]
De hecho, un estudio en Suecia mostró que las restauraciones directas en pacientes con casos severos de AI o con formas hipomineralizadas / hipomaturas de AI duran menos que en pacientes con casos moderados de AI o formas hipoplásicas de AI, aunque los los datos recogidos en investigaciones de laboratorio muestran que las diferencias observadas resultan de las variaciones en el el diseño empleado y la gravedad de AI en lugar de las diferencias en las características histológicas del esmalte afectado, o sea del tipo de AI. Para la rehabilitación con coronas, el tipo de AI no parece ser importante. Además, las restauraciones de cerámica total presentan una longevidad similar a las restauraciones de aleaciones o aleaciones chapeadas.[9]
Las restauraciones directas con compuestos a base de resinas son comúnmente preferidas en pacientes jóvenes para evitar la preparación extensa de los dientes durante la adolescencia; Sin embargo, los datos más recientes indican que las restauraciones indirectas duran más que las restauraciones directas, lo que se debe a la unión imperfecta de los ligamentos al esmalte afectado por la AI. Aunque las complicaciones de endodoncia se observan con poca frecuencia, la rehabilitación de los pacientes con AI que emplean restauraciones indirectas debe comenzar tan pronto como sea posible.[9]
El tratamiento de los tipos de amelogénesis imperfecta con esmalte severamente afectado requiere múltiples fases para mantener la forma, función y estética de la dentición primaria (2-6 años de edad), de la dentición mixta que se observa en la niñez (6-11 años) y de la dentición permanente (11 años o mayores). El cuidado restaurativo inicial debe comenzar con la prevención en la dentición primaria considerando todos los aspectos.[5] La intervención temprana debe comenzar con acciones preventivas que incluye, la instrucción de higiene oral, la aplicación tópica de flúor, la aplicación de sellantes, enjuagues con clorhexidina, dentífricos desensibilizantes y extracción de dientes que tienen un mal pronóstico. Para recuperar los dientes se puede hacer restauraciones con resinas directas e indirectas y coronas de metal y/o de cerámica como anteriormente comentado.[6][10]
El tratamiento puede requerir la intervención de múltiplos especialistas. La mala oclusión frecuentemente requiere intervención de ortodoncia y también quirúrgica. El tratamiento puede durar varias décadas ya que incluye la dentición primaria y permanente. El conocimiento de los defectos genéticos asociados con los tipos específicos de amelogénesis está aumentando lo que lleva a una mejora del diagnóstico del tipo especifico. Cuando se sabe el problema genético es mejor porque permite predecir que pacientes podrán tener problemas asociados como formación de placa y mala oclusión y así escoger el mejor plano de tratamiento.[5]
Última actualización: 6/12/2018
Los grupos de apoyo y las organizaciones de ayuda pueden ser de utilidad para conectarse con otros pacientes y familias, y pueden proporcionar servicios valiosos. Muchos proporcionan información centrada en el paciente, e impulsionan la investigación para desarrollar mejores tratamientos y para encontrar posibles curas. Pueden ayudar a encontrar estudios de investigación, y otros recursos y servicios relevantes. Muchas organizaciones también tienen asesores medicos expertos o pueden proporcionar listas de médicos y/o clínicas. Visite el sitio en la red del grupo que le interese o póngase en contacto con ellos para conocer los servicios que ofrecen. Recuerde que la inclusión en esta lista no representa un aval de GARD.
Sitios o Redes Sociales en la Internet
-
La Federación Chilena de Enfermedades Raras (FECHER) es un grupo de apoyo en Facebook para los afectados con enfermedades raras.
Organizaciones de Apoyo General
-
Alianza Iberoamericana de Enfermedades Poco Frecuentes
Correo electrónico: aliber@aliber.org
Enlace en la red: http://aliber.org/ -
American Academy of Pediatric Dentistry (AAPD)
211 East Chicago Avenue, Suite 1600
Chicago, IL 60611-2637
Teléfono: (312) 337-2169
Fax: (312) 337-6329
Correo electrónico: http://www.aapd.org/contact/
Enlace en la red: http://www.aapd.org/ -
Asociación Todos Unidos Enfermedades Raras Uruguay (ATUERU)
Emilio Castelar 440 esquina Pirán Torre CH
Apto 502- Malvín Norte
Montevideo, Uruguay
Teléfono: (598) 2 5227328
Correo electrónico: atueru.eerr@gmail.com
Enlace en la red: http://atueru.org.uy/ -
EURORDIS Plateforme Maladies Rares
96, rue Didot
75014 Paris
France
Teléfono: 33 1 56 53 52 10
Fax: 33 1 56 53 52 15
Correo electrónico: eurordis@eurordis.org
Enlace en la red: http://www.eurordis.org -
Federación Argentina de Enfermedades Poco Frecuentes (FADEPOF)
Argentina
Correo electrónico: info@fadepof.org.ar
Enlace en la red: http://fadepof.org.ar -
Federación Colombiana de Enfermedades Raras (FECOER)
Av. Cra 15 #124 -17 Edificio Jorge Barón Torre B Oficina 703
Bogotá, Colombia
Teléfono: 320 944 5674
Correo electrónico: info@fecoer.org
Enlace en la red: http://www.fecoer.org -
Federación Española de Enfermedades Raras (FEDER)
C/ Pamplona 32 – CP: 28039 Madrid
España
Teléfono: 915334008
Correo electrónico: feder@enfermedades-raras.org
Enlace en la red: http://www.enfermedades-raras.org -
Federación Mexicana de Enfermedades Raras (FEMEXER)
Correo electrónico: info@femexer.org , femexer@gmail.com
Enlace en la red: http://www.femexer.org/ -
Federación Peruana de Enfermedades Raras (FEPER)
Lima, Perú
Teléfono: 511 795 0304
Correo electrónico: enfermedadesrarasperu@gmail.com
Enlace en la red: http://feperperu.blogspot.com.es/
Contact Form - http://feperperu.blogspot.com/p/contacto.html -
Fundación Geiser
Grupo de Enlace, Investigación y Soporte para Enfermedades Raras
Nicolás Avellaneda 595
Mendoza, Argentina/ Serrano, 669
Ciudad Autónoma de Buenos Aires
Argentina
Teléfono: 54 261 429-1987
Correo electrónico: info@fundaciongeiser.org
Enlace en la red: https://www.facebook.com/fundaciongeiser.enfermedadesraras/ -
Organización Mexicana de Enfermedades Raras (OMER)
Correo electrónico: info@omer.org.mx
Enlace en la red: http://omer.org.mx/
¿Conoce alguna organización? ¡Nos gustaría recibir sus comentarios!
Estos recursos proporcionan más información sobre esta condición o de los síntomas asociados. Los recursos en la sección “Información detallada” contiene lenguaje médico y científico que puede ser difícil de entender. Es posible que desee revisar esta información con un médico.
Comience por aquí
-
Más información sobre esta enfermedad está disponible en Genetics Home Reference (GHR), el sitio Web de la National Library of Medicine (NLM) de los Estados Unidos para información al consumidor sobre enfermedades genéticas y los genes o cromosomas relacionados a esas enfermedades. Este recurso también provee descripciones de conceptos genéticos, un glosario de términos genéticos, y enlaces a otras fuentes de información sobre la genética. (en inglés)
-
MedlinePlus brinda información detallada, confiable y actualizada acerca de esta enfermedad, su señales, o sus síntomas en un lenguaje fácil de leer. Visite el enlace para leer sobre la(s) causa(s), los síntomas, pruebas y exámenes, tratamiento, pronóstico, y más.
Información Detallada
-
Orphanet es una base de datos europea de aceso gratuito en la red sobre enfermedades raras y medicamentos huérfanos. Contiene enciclopedias médicas y un directorio de servicios especializados como servicios médicos, laboratorios, proyectos de investigación y asociaciones de pacientes.
Preguntas recibidas por GARD pueden ser publicadas si se considera que la información podría ser de utilidad para otras personas. Antes de publicar una pregunta se elimina toda la información de identificación para proteger su privacidad. Si no desea que su pregunta sea publicada, por favor díganos.
¿Tiene alguna pregunta? Póngase en contacto con un especialista en información de GARD.
-
Rosemberg J. Amelogenesis imperfecta. Medline. 2018; http://www.nlm.nih.gov/medlineplus/spanish/ency/article/001578.htm.
-
Amelogenesis imperfecta. Genetics Home Reference (GHR). May 2015; http://ghr.nlm.nih.gov/condition/amelogenesis-imperfecta.
-
Amelogenesis Imperfecta. National Organization for Rare Diseases (NORD). 2018; https://rarediseases.org/rare-diseases/amelogenesis-imperfecta/.
-
Wright JT. Amelogenesis Imperfecta. Developmental Defects of the Teeth. https://www.dentistry.unc.edu/dentalprofessionals/resources/defects/ai/.
-
Wright JT. Development Defects of the Teeth. UNC Dental Research. https://www.dentistry.unc.edu/dentalprofessionals/resources/defects/ai/.
-
Gonzales Pinedo CO & Perona-Miguel de Priego G. Amelogenésis imperfecta: Criterios de clasificación y aspectos genéticos. Rev Estomatol Herediana. 2009; 19(1):55-62. http://www.upch.edu.pe/faest/publica/2009/vol19_n1/vol19_n1_09_art9.pdf.
-
Leung VW, Low B, Yang Y & Botelho MG. Oral Rehabilitation of Young Adult with Amelogenesis Imperfecta. J Contemp Dent Pract. May 1, 2018; 19(5):599-604. https://www.ncbi.nlm.nih.gov/pubmed/29807973.
-
Crawford PJM, Aldred M & Bloch-Zupan A. Amelogenesis imperfecta. Orphanet. April, 2007; http://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=88661.
-
Strauch S & Hahnel S. Restorative Treatment in Patients with Amelogenesis Imperfecta: A Review. J Prosthodont. January 29, 2018; https://www.ncbi.nlm.nih.gov/pubmed/29377372.
-
Wright JT. The molecular etiologies and associated phenotypes of amelogenesis imperfecta. Am J Med Genet. 2006; 140A:2547-55. http://ghr.nlm.nih.gov/condition/amelogenesis-imperfecta.
¿Conoce algún artículo de revisión? ¡Nos gustaría recibir sus come
Fuente: https://rarediseases.info.nih.gov/espanol/12002/amelogenesis-imperfecta
Amelogenesis_imperfecta
La amelogenesis imperfecta es una enfermedad genética que se presenta con formación anormal del esmalte o capa externa de los dientes. El esmalte está compuesto principalmente por mineral, que es formado y regulado por las proteínas en él. La amelogenesis imperfecta es debida al mal funcionamiento de las proteínas en el esmalte: ameloblastina, enamelina, tuftelina y amelogenina.
Las personas afectadas con amelogenesis imperfecta tienen dientes con color anormal: amarillo, marrón o gris. Los dientes tienen un mayor riesgo de sufrir cavidades dentales y son hipersensitivos a los cambios de temperatura. Este desorden puede afectar cualquier número de dientes.
Genética[editar]
Hasta la fecha, mutaciones en los genes: AMELX, ENAM, MMP20, y KLK-4 han sido encontrados de causar amelogenesis imperfecta (forma no-sindrómica).
Los genes AMELX, ENAM, KLK-4 y MMP20 proveen instrucciones para producir proteínas que son esenciales para el normal desarrollo de los dientes. Estas proteínas están involucradas en la formación del esmalte, el cual es duro, material rico en calcio que forma la capa exterior de cada diente.
Las mutaciones en algunos de estos genes alteran la estructura de estas proteínas o incluso impiden a los genes producir por completo las proteínas. Como resultado el esmalte dental es anormalmente fino o suave y puede tener un color amarillo o marrón. Los dientes con esmalte defectuoso son débiles y fácilmente dañados.
Los investigadores están buscando mutaciones en otros genes que puedan también causar amelogenesis imperfecta.
La amelogenesis imperfecta puede tener diferentes patrones de herencia dependiendo del gen que es alterado. La mayoría de los casos son causados por mutaciones en el gen ENAM, y son heredados con un patrón autosómico dominante. Este tipo de herencia significa que una copia del gen alterado en cada célula es suficiente para causar el desorden.
La amelogenesis imperfecta es también heredada en un patrón autosómico recesivo; esta forma de desorden puede resultar de mutaciones en el gen ENAM or MMP20. Una herencia autosómico recesiva significa que dos copias del gen en cada célula están alteradas.
Cerca del 5% de los casos de amelogenesis imperfecta son causadas por mutaciones en el gen AMELX y están vinculadas a un patrón de enlace X. Una condición se considera enlazada X si el gen mutado que causa el desorden se localiza en el cromosoma X, uno de los dos cromosomas sexuales. En muchos casos, el macho con una forma enlazada X de esta condición experimenta más severas anormalidades dentales que una hembra afectada.
Otros casos de esta condición resultan de nuevas mutaciones de genes y ocurren en gente sin historia de desórdenes genéticos en su familia. así pues la amelogenesis imperfecta muta nuevamente.
Tratamiento[editar]
Las directrices actuales para el tratamiento de restauración sugieren que se debe cubrir la superficie dental con composite directo o carillas de composite hasta la edad adulta y se recomiendan las coronas de acero inoxidable para los primeros molares permanentes como una solución temporal en la infancia y adolescencia.
Los pacientes con Amelogénesis Imperfecta a menudo piden un tratamiento más permanente en una edad temprana.[1]
Referencias[editar]
-
↑ Pousette Lundgren, Gunilla; Karsten, Agneta; Dahllöf, Göran (10 de diciembre de 2015). «Oral health-related quality of life before and after crown therapy in young patients with amelogenesis imperfecta». Health and Quality of Life Outcomes (en inglés) 13 (1). PMC 4676094. PMID 26651486. doi:10.1186/s12955-015-0393-3. Consultado el 3 de marzo de 2016.
<img src="//es.wikipedia.org/wiki/Special:CentralAutoLogin/start?type=1x1" alt="" title="" width="1" height="1" style="border: none; position: absolute;" />
Obtenido de «https://es.wikipedia.org/w/index.php?title=Amelogenesis_imperfecta&oldid=113247346»
-
Esta página se editó por última vez el 12 ene 2019 a las 23:03.
-
El texto está disponible bajo la Licencia Creative Commons Atribución Compartir Igual 3.0; pueden aplicarse cláusulas adicionales. Al usar este sitio, usted acepta nuestros términos de uso y nuestra política de privacidad.
Wikipedia® es una marca registrada de la Fundación Wikimedia, Inc., una organización sin ánimo de lucro
Fuente: https://es.wikipedia.org/wiki/Amelogenesis_imperfecta
Diagnóstico y tratamiento integral en pacientes con Amelogénesis Imperfecta
Reporte de un caso
María del Carmen López Jordi1, Esther Szwarc2
Resumen
Objetivo: La Amelogénesis Imperfecta comprende un grupo heterogéneo de defectos del esmalte de origen genético, debidos a alteraciones en la formación del esmalte dentario, en calidad y/o cantidad. El diagnóstico se basa en la observación clínica, exámenes radiográficos, la historia familiar, el árbol genealógico y cuando es posible el diagnóstico genético. Se caracteriza por tener un amplio rango de presentaciones clínicas en ambas denticiones. Esta afección tiene un alto impacto en niños y adolescentes debido a que la carencia estética y la disfunción limitan su calidad de vida. La atención integral se convierte en un aspecto esencial y demanda una inteligente y necesaria interacción profesional, paciente y familia, la cual debe establecerse en forma temprana y de manera interdisciplinaria. Objetivo: Presentar un reporte de casocaso de un paciente de 11 años con Amelogénesis Imperfecta y diagnóstico clínico y radiográfico de tipo hipoplásico, apoyado en su historia familiar. El tratamiento integró varias etapas: uso de agentes remineralizantes a fin de restaurar los tejidos dentarios; ortodoncia para crear espacio para la erupción del canino retenido (13) y alineación de la arcada dentaria superior y rehabilitación dentaria con resinas compuestas y coronas metálicas fenestradas en oclusal. Conclusiones: El seguimiento por cinco años con una actitud muy positiva de la paciente hacia el mantenimiento de su salud, confirma que en el adolescente, una sonrisa saludable es importante en el desarrollo de la autoestima y las relaciones interpersonales.
Palabras clave: Salud bucal, Amelogénesis Imperfecta, genética, atención odontológica interdisciplinaria.
Relato de um caso
Diagnóstico e tratamento integral em pacientes com Amelogênese Imperfeita. Relato de um caso
Resumo
A Amelogênese Imperfeita compreende um grupo heterogêneo de defeitos de esmalte de origem genética, que ocorre devido a alterações na formação de esmalte dentário, e podendo comprometer em qualidade e/ou quantidade do mesmo. O diagnóstico é baseado em observação clínica e radiográfica, história familiar, padrão genético familiar e quando possível, realização de uma investigação genética. Ela caracteriza-se por ter uma ampla gama de manifestações clínicas em ambas as dentições. Esta condição tem um alto impacto em crianças e em adolescentes, gerando um comprometimento social, a funçáo e consequentemente, limitando a qualidade de vida dos pacientes. O cuidado integral torna-se um aspecto essencial do tratamento, exigindo uma interação do profissional com o paciente e seus familiares, que deve ser estabelecida de forma precoce e interdisciplinar. Os objetivos do plano de tratamento devem abranger três aspectos: prevenção, restauração da estrutura dental e reabilitação estética. Objetivo: Paciente de 11 anos de idade, com amelogênese Imperfeita de tipo hipoplásico, segundo o diagnostico clínico e radiográfico e com base na história familiar. O tratamento foi realizado em diversas etapas: uso de agentes remineralizantes na remineralizacão de tecidos dentais, tratamento ortodóntico para criar espaço para a erupção do canino retido e o alinhamento da arcada dentária superior, e por último, a reabilitação com resinas compostas e coroas metálicas fenestradas na superfície oclusal. Conclusão: Durante cinco anos de acompanhamento, o paciente tem demonstrado uma atitude muito positiva em relação à manutenção de sua saúde, confirmando-se que na adolescência, um sorriso saudável é importante no desenvolvimento da autoestima e das relações interpessoais.
Palavra-chave: Saúde bucal, Amelogênese Imperfeita, genética, assistência odontológica integral.
Case report
Diagnosis and comprehensive treatment for patients with Amelogenesis Imperfecta. Case report
Abstract
Amelogenesis Imperfecta is a diverse group of hereditary and heterogeneous enamel defects, due to alterations in the formation of dental enamel in quality and/or quantity. Diagnosis is based on clinical and radiological findings, family history, family tree, and genetic diagnosis when it is possible. It is characterized by a wide range of clinical presentations in both dentitions. This condition has a high impact on children and adolescents, generates a very disadvantageous social performance since aesthetic problems and dysfunction limit their quality of life. Comprehensive care becomes an essential aspect and it demands a close and necessary professional, patient and family interaction, which must be established early and in an interdisciplinary way. Objective: We present a patient with Amelogenesis Imperfecta, 11 years old, with a clinical and radiographic diagnosis of hypoplastic type, based on her family history. The treatment integrated several stages: use of remineralizing agents in order to restore Latinoamericanadental tissues, orthodontics to create space for the eruption of the retained canine (13), and the alignment of upper dental arch, and rehabilitation with composite resins and metal crowns fenestrated in occlusal. Conclusion: The five year follow- up, with a very positive attitude of the patient toward the maintenance of her health, suggests that in adolescence, a healthy smile is important in the development of self-esteem and interpersonal relationships.
Key words: Oral health, Amelogenesis Imperfecta, genetics, comprehensive dental care.
-
Profesora Mg. Directora Maestría en Ciencias Odontológicas-opción Odontopediatría. Facultad de Odontología. Universidad de la República. Uruguay.
-
Profesor Agregado. Especialista en Odontopediatría. Facultad de Odontología. Universidad de la República. Uruguay.
Introducción
En los últimos años se han logrado importantes avances en la comprensión de los mecanismos biológicos y etiológicos de las enfermedades orales, así como en el desarrollo de terapéuticas efectivas. Hoy día surge un nuevo paradigma de salud, el biopsicosocial, con implicaciones tanto para la clínica, la investigación y las políticas de salud. Este nuevo paradigma agrega a la salud y su atención, el concepto de calidad de vida y el reconocimiento de los determinantes funcionales y sociales sobre la misma. La Federación Dental Internacional (FDI), en el marco de su Asamblea General 2016, propone una nueva definición de Salud Bucal, como concepto dinámico, de naturaleza subjetiva y muy influenciada por el pasado social y cultural de la persona1,2 “La salud oral es multifacética e incluye la habilidad para hablar, reír, oler, saborear, tocar, masticar, tragar y transferir una serie de emociones a través de las expresiones faciales con confianza y sin dolor, incomodidad ni enfermedad del complejo craneofacial”. La mencionada definición y sus componentes son pertinentes a la problemática que motiva este trabajo, cuando las personas ven su desempeño social perjudicado ya que una disfunción sumada a una carencia estética limita su calidad de vida.
La Amelogénesis Imperfecta (AI) comprende un grupo heterogéneo de defectos del esmalte de origen genético, con alteraciones en el proceso de formación del tejido adamantino, en su calidad y/o la calidad.3 La prevalencia de AI varía geográficamente: de 1/700 en Suecia a 1/14.000 en Estados Unidos, afecta a ambos sexos y a dientes temporarios y permanentes.4 La AI presenta distintos patrones hereditarios, puede transmitirse a partir de un patrón autosómico dominante (AD), por uno recesivo (AR), ligado al cromosoma X o aún por mutación genética espontánea.5 Los principales genes candidatos a mutaciones causales de AI, son aquellos que codifican proteínas involucradas en la ruta de formación del esmalte, modulando la deposición mineral y el crecimiento del órgano del esmalte (AI no sindromática). Asimismo, pueden verse afectados otros tejidos orales y extra orales (AI sindromática). La complejidad del patrón de enfermedad sugiere la ocurrencia de mutaciones en más de 15 genes en la etiología de AI no sindromáticas siendo los más conocidos: amelogenina, enamelina, ameloblastina, tuftelina y amelotina.3,6 Una reciente investigación identifica que en la ameloblastina se encuentra la más importante mutación y al ser la encargada de la generación de la primera fase del desarrollo del esmalte (secretora), su mutación genera un daño dental profundo. La segunda mutación descubierta es de la amelotina producida en la última fase de maduración del esmalte, cuando este debe calcificarse y endurecerse.7
El diagnóstico de AI se basa habitualmente, en la observación clínica, la historia familiar, el árbol genealógico y exámenes paraclínicos. La radiografía panorámica puede dar los primeros indicios: defectos en el tamaño dentario, presencia de dientes retenidos etc. y las periapicales aportan mayor precisión respecto a: trastornos de la mineralización, falta de contraste entre esmalte y dentina, y tamaño de cámara pulpar. Actualmente, el diagnóstico genético no se puede aplicar sistemáticamente, siendo un recurso muy importante para la investigación. La AI se caracteriza por tener un amplio rango de presentaciones clínicas (fenotipos).9 Varios sistemas de clasificación han sido propuestos basados en: la herencia, la histopatología y las características dentarias específicas. La clasificación de Witkop 4 distingue cuatro tipos de AI basándose en el fenotipo que pueden subdividirse en 15 categorías en función del patrón hereditario. Recientemente, se propuso que la herencia sea el principal factor de clasificación dando lugar a una nueva clasificación propuesta por Neville et al. (2016)10 a) Amelogénesis imperfecta hipoplásica, b) Amelogénesis imperfecta hipocalcificada, con sus variantes hipomaduración e hipocalcificación y c) Amelogénesis imperfecta con taurodontismo (AI hipomadura/hipoplásica). Se propone avanzar hacia un nuevo sistema de clasificación, basado en el modo de herencia, con discriminantes secundarios que incluyan fenotipo, bases moleculares y resultados bioquímicos, que en el futuro serán de directa aplicación en el tratamiento del paciente.10 El diagnóstico diferencial de AI con otros trastornos del esmalte no es sencillo dada la inespecificidad de algunas de las manifestaciones siendo importante distinguir la fluorosis dental, la hipomineralización molar-incisiva (MIH) y otras displasias del esmalte no generalizadas. La AI se ha asociado con diferentes alteraciones dentarias (discoloraciones, microdoncia, agenesias) y también con maloclusiones (mordida abierta y mordida cruzada) que pueden resultar de alteraciones en los mecanismos eruptivos, hábitos de succión y pérdida de dimensión vertical.11 Los diferentes autores reconocen que la mayor queja de los pacientes afectados de AI, son la insatisfacción estética y la alta sensibilidad dentaria, lo que obliga a abordarlo con una visión integral. Esta condición se acompaña de problemas psicológicos y ausencia de seguridad, debido a la apariencia de los órganos dentarios.12 Autores como Coffield et al.13 señalan que el impacto originado por ser portador de AI, es comparable al generado por condiciones sistémicas que afectan severamente la salud. Esto implica para el paciente, deterioro de la percepción de su imagen, autoestima y confianza. La carencia estética impone al individuo retos sociales de interacciones e incluso auto percepción negativa.14-15 Las anomalías dentarias, presentes en la AI, colocan a quienes la poseen en desventaja psicosocial y en definitiva perjudica su calidad de vida. Por ser una afección que se presenta desde la erupción de los dientes e interferir en tantos campos de la salud, la atención integral debe brindarse en forma temprana reconociéndose como un factor clave en la calidad de la misma.16
En el tratamiento influyen factores como: edad, tipo y grado de severidad de la afección, situación intraoral, otros factores de riesgo, contexto socioeconómico y cultural etc. El diagnóstico temprano asegura un plan de tratamiento adecuado cubriendo tres aspectos: prevención, restauración tisular-dentaria y rehabilitación estética. Es importante motivar al paciente y sus padres hacia una completa rehabilitación bucal. El plan de tratamiento preventivo es esencial debido a aspectos funcionales y para lograr un impacto psicológico positivo, incluye: instrucciones de higiene oral, utilización sistemática de dentífricos fluorados y consejos dietarios. La higiene oral puede resultar dificultosa en pacientes con AI por sensibilidad durante el cepillado por lo que se recomienda un cepillo de cerdas suaves y agua tibia para el enjuague lo cual mejora los síntomas y la efectividad del procedimiento. Actualmente los productos que potencian la restauración tisular promoviendo su remineralización tienen muy buena receptividad en la profesión y sus éxitos clínicos son evidentes. Tal es el caso del complejo de Caseín Fosfopéptido y Fosfato de Calcio Amorfo (CPP-ACP) comercializado como Recaldent™ (GC America) incluido cremas para aplicación tópica y gomas de mascar que promueven la remineralización dentaria. En condiciones de pH neutro hay una disolución de los cristales de hidroxiapatita, liberando iones de calcio, fosfato e hidroxilo al fluido oral que conduce a una sobresaturación de la solución precipitando los minerales nuevamente en la superficie dentaria lo que puede considerarse una remineralización. Se ha demostrado que ese incremento de iones biodisponibles lo proporciona la aplicación con ACP-CPP uno o dos veces al día durante 10 días.17 Con la aplicación de cremas que contienen CPP-ACP, los péptidos son capaces de interactuar con las proteínas salivales resultando esenciales durante el proceso de remineralización.18 Por otro lado, el CPP-ACP es útil en reducir la hipersensibilidad dentaria produciendo la oclusión de los túbulos dentinarios. En pacientes con AI mayores de seis años se indica la presentación que combina caseín fosfopéptido-fosfato de calcio amorfo con fluoruro (900 ppm F-), representado por CPP-ACFP (MI Paste Plus) que según Reynolds (2008)19 genera un incremento de iones calcio y fosfato biodisponibles. Esta acción sinérgica aumenta la incorporación del fluoruro resultando en una mayor mineralización del esmalte sub-superficial comparado con la utilización del fluoruro solo. Un beneficio adicional de este compuesto, lo constituye la mejora en la apariencia estética de las opacidades, según. Reynolds una mayor concentración de calcio, fosfato y fluoruro en la superficie adamantina conduce a su difusión en el espesor de todo el esmalte, resultando en niveles más altos y homogéneos de mineralización.20
Luego de culminada la etapa rehabilitadora, el paciente debe seguir cumpliendo un plan preventivo importante para el éxito a largo plazo de las restauraciones estéticas ya que requerirá tratamiento adicional, reparador, a intervalos variables a fin de mantener el cierre marginal como parte del mantenimiento de la salud.21 El tratamiento rehabilitador inicial, debe ser conservador, respetando lo más posible los tejidos dentarios utilizando para las restauraciones cementos de ionómero de vidrio de alta densidad o combinados con resinas de fotocurado. En la medida que estas restauraciones se mantienen y la sensibilidad se reduce en forma considerable, se comienza a realizar restauraciones de resina compuesta logrando una mejor estética y durabilidad. Es importante lograr un buen grabado dado que hay un alto porcentaje de fallas en la adhesión de las restauraciones en pacientes con AI especialmente en el tipo de amelogénesis con hipocalcificación.
Caso clínico
Paciente de 10 años de edad, sexo femenino, consulta en 2012, a la Carrera de Especialización en Odontopediatria, Facultad de Odontología, Universidad de la República, Uruguay. En la Historia Clínica Institucional está registrado en forma expresa el Consentimiento Informado firmado por la madre de la paciente luego de recibir ambas información adecuada, suficiente y continua para los procedimientos diagnósticos, terapéuticos, registro fotográfico y su eventual publicación. El motivo de consulta es la falta de estética de sus dientes, que según relato del paciente y su familia, comenzó con la erupción de dientes permanentes y también gran sensibilidad dentaria frente a alimentos, bebidas y cepillado bucal. La historia describe que su nacimiento fue por parto natural presentando pautas de crecimiento y desarrollo normales. Al momento de la consulta en nuestra Clínica, se observa que la niña presenta un desarrollo físico-psico-social de acuerdo a su edad cronológica, sistémicamente sana (controlada regularmente en salud) y no se encuentra bajo tratamiento médico.
Antecedentes odontológicos
El tiempo de erupción de la dentición temporaria y mixta fue normal y relata experiencia de caries en los dientes caducos. Su dieta es cariogénica con alto contenido de jugos y alfajores. Su experiencia odontológica fue buena y es derivada a la Facultad por los profesionales tratantes para establecer un diagnóstico de su condición. Presenta múltiples restauraciones con Ionómero de vidrio y aparatología ortopédica (se retira al ingresar a la Especialidad).
Antecedentes familiares
Bisabuela paterna, tío abuelo y padre presentaron la misma condición bucal (dientes oscuros y pequeños), posee un hermano menor odontológicamente sano. Actualmente el padre es portador de prótesis completa. Se aprecia un entorno familiar con buen clima educativo y social y cumplidor en sus consultas.
Historia actual
La paciente relata alta sensibilidad a los cambios térmicos que le dificulta la higiene y la alimentación. También informa sobre sus problemas estéticos, se advierte que no quiere mostrar los dientes por lo que cuesta que sonría y temor al examen bucal por miedo a sentir dolor al contacto con instrumentos, aire o agua.
Examen clínico y radiográfico 2012 (Figuras 1-3)
Presenta 27 dientes permanentes erupcionados, Índice de Higiene Oral Simplificado (HIOS) de 1.7, Índice de caries/diente (CPOD) de 6, Índice de caries/superficie (CPOS) de 13, no erupción del canino superior derecho, alteraciones en estructura del esmalte, dientes de color amarillento, bordes incisivos atricionados, ausencia de puntos de contacto y superficies de aspecto relativamente rugoso, restauraciones plásticas en: 14, 16, 26, 36, 44 y 46 (defectuosas), línea media desviada a la derecha, “overjet” y “overbite” de apariencia normal. Al examen funcional: respiración nasal, fonación normal, deglución adulta y masticación bilateral y alternada. Radiográficamente se aprecia el número de dientes correcto, el 13 retenido y el germen del 48, desarrollo dentario y erupción normal y se identifica que no existe otra patología ósea ni malformaciones salvo en el espesor de los tejidos dentarios, muy fino en algunas piezas, pero con contraste casi normal del esmalte con la dentina.
Figura 1. Examen clínico 2012. Figura 2. Examen clínico 2012. Figura 3. Ortopantomografía 2012.
Diagnóstico
Amelogénesis Imperfecta (leve) no sindrómica, de tipo hipoplásica. La etiología hereditaria podría determinarse probablemente ligada al cromosoma X o de tipo autosómica dominante de acuerdo a las características de su árbol genealógico. Tratamiento 2012-2014, 2015: 1) TERAPIA BÁSICA: Enseñanza de higiene oral con cepillo blando, dentífrico fluorado 1.500 ppm y enjuague bucal con agua tibia, Indicación de MI Paste Plus dos veces al día, remotivación a la paciente y familia en cada sesión; control de biofilm y control de dieta. 2) TERAPIA REHABILITADORA: Sellado con Ionómero de vidrio modificado con resina en las zonas dentarias hipoplásicas (17, 15, 14, 13, 24, 25, 26, 27, 37, 35, 34, 33, 43, 44, 45, 47), coronas de acero fenestradas en oclusal y resinas en 16, 26, 36, 46 (Figuras 4 y 5), controles periódicos estrictos (semanales y quincenales). La telerradiografía (Figura 6) y el cefalograma muestran que es una paciente en crecimiento, con perfil recto, crecimiento rotacional posterior (dólico-facial suave), Clase I molar, Clase II ósea, “overbite y overjet” disminuido (tendencia a mordida abierta), 13 retenido, ángulo interincisivo aumentado e incisivo inferior levemente retruido lo que llevó a la colocación de aparatología ortodóncica en el 2015, a fin de crear espacio para el canino, y alinear la arcada superior. (Figura 7).
Figura 4. Maxilar superior 2015. Figura 5. Maxilar inferior 2015. Figura 6. Telerradiografía. Figura 7. Aparatología ortodóntica 2016.
Tratamiento 2017
Retiro de aparatología ortodóncica. 1) terapia básica: Remotivación a la paciente y su familia, control de biofilm, dentífrico fluorado 1500 ppm, aplicación domiciliaria de MI Paste Plus 2 veces al día, a fin de remineralizar las lesiones blancas que se evidencian al retiro de los brackets. 2) terapia rehabilitadora: restauración de todos los dientes con resina de fotocurado, controles quincenales estrictos (Figuras 8, 9). Seguimiento del desarrollo de terceros molares e interconsulta con equipo interdisciplinario, aparatología ortodóncica y rehabilitación definitiva.
Figura 8. Control clínico 2017. Figura 9. Control radiográfico 2017.
Discusión
La AI constituye un grupo de afecciones heterogéneas tanto clínica como genéticamente, caracterizadas por defectos en el esmalte. En nuestro país, no existen estudios de prevalencia respecto a dicha patología. Habitualmente, se derivan a la Especialidad de Odontopediatría de nuestra Facultad, por ser un centro de referencia a nivel nacional, casos con patologías de formación de tejidos dentarios, lo cual puede darnos una falsa idea de su frecuencia. La paciente que presentamos, fue derivada en su etapa de dentición mixta con la finalidad de establecer un diagnóstico que explicara la falta de estética de los dientes permanentes recién erupcionados y su gran sensibilidad. Nuestro diagnóstico primario, fue AI de tipo hipoplásico.
Dada la importancia de la herencia en su etiología y transmisión, los estudios genéticos son la mejor base para un diagnóstico certero de esta afección. Recientemente, a través de estudios clínicos, genéticos y moleculares de familias afectadas de AI, se están estableciendo correlaciones feno-genotípicas en este grupo de anomalías. Se espera que los mismos colaboren en un mayor conocimiento de la afección para que puedan contribuir a mejorar su pronóstico y faciliten su aplicación. Debido a las dificultades tecnológicas disponibles en nuestro medio y la escasa posibilidad de acceso a exámenes de ese tipo, la anamnesis, el examen clínico y radiográfico continúan siendo los más utilizados para establecer su diagnóstico. La AI de tipo hipoplásica, se caracteriza por inadecuada deposición de matriz del esmalte, en cambio la mineralización, cualquiera sea la cantidad de matriz presente, se produce en forma adecuada y con apropiado contraste radiográfico del esmalte con la dentina subyacente. De acuerdo a la clasificación de Neville, nuestra paciente presenta una AI de tipo hipoplásico con mayor semejanza a la de patrón punteado generalizado. Las fositas se encuentran desparramadas a lo largo de la superficie dentaria y no se correlacionan con un patrón de alteración medioambiental. El esmalte entre las fosas es de espesor, dureza y coloración normal, presenta atrición ligera de bordes incisales y falta más marcada de esmalte en la superficie oclusal, no característica de un patrón localizado. Los aspectos fenotípicas de la paciente, de acuerdo a la citada clasificación, mostraría correspondencia con el tipo de herencia autosómica dominante. Dado que, en el árbol genealógico de la paciente se relata como antecedente más remoto haber sido presentada por su bisabuela paterna, un tío abuelo (que no lo transmitió a su descendencia y solo tuvo hijos varones) y su padre, se evaluó que también podría ser ligada al cromosoma X, ya que esta forma no se transfiere de individuos masculinos a masculinos y todas las hijas del hombre afectado son portadoras del defecto genético. (La paciente es única hija mujer del matrimonio y solo tiene un hermano que no presenta AI). El no recordar otros antecedentes del árbol genealógico y la no realización de exámenes genéticos, mantienen dichos diagnósticos como hipotéticos.
Otro dato a resaltar es que según la paciente y su familia, no presentó AI en la dentición caduca (relata experiencia de caries). Dado que se resalta en la literatura que AI afecta ambas denticiones, deducimos que debido al reducido espesor de esmalte en la dentición primaria (menor discoloración) se pudo enmascarar dicha patología y no ser diagnosticada tempranamente por el odontólogo. Las diferentes anomalías dentarias como ser: microdoncia, ausencias congénitas, calcificaciones pulpares e, hipercementosis no están presentes.8,11 Se constató un canino permanente superior retenido y ausencia de algunos puntos de contacto por desgastes e incisivos atricionados pero no se asoció a mordida abierta (aunque hay tendencia a ella), sin pérdida de dimensión vertical. No presentó alteraciones gingivales de consideración, salvo en el inicio del tratamiento una ligera gingivitis relacionada a biopelícula, por defecto de cepillado.
En cuanto al tratamiento, incluyó todas las etapas que clásicamente describe la literatura. En la fase preventiva, debemos destacar, la positiva respuesta tanto de la paciente, como de su familia. Fue fundamental el empleo, como agente remineralizante, del complejo CPP-ACP (Recaldent TM GG-América).17-20 Su uso adecuado, provocó un rápido descenso de la hipersensibilidad dentinaria, el cual se demostró en el descenso del índice de higiene oral simplificado de 1.7 a 0.6 en las primeras consultas. También es de destacar la modificación de la dieta cariogénica que presentaba por otra más saludable, a pesar de estar transitando la adolescencia. La etapa rehabilitadora fue adaptada en forma individual a las particularidades de la paciente. Se comenzó con restauraciones con vidrio ionómero y se fueron aportando soluciones más estéticas y funcionales de acuerdo a la respuesta y crecimiento de la paciente. Es de destacar, que en la rehabilitación de los molares 16-26-36 y 46, se optó por coronas de acero fenestradas en oclusal y resinas. Como ventajas de su utilización, señalamos no solamente restituir puntos de contacto proximales, sino también, colaborar a no alterar más la dimensión vertical permitiendo un mejor control de los contactos en relación céntrica y excéntrica comparados con las coronas metálicas totales. En el sector anterior se utilizaron resinas, con buenos adhesivos dentinarios, procurando soluciones cada vez más estéticas, no teniendo problemas con la adhesión, tanto de las restauraciones como con los brackets para la reubicación del canino retenido y posterior alineación de la arcada superior, a pesar de que se relatan habituales fallas de grabados principalmente en los casos de AI del tipo hipocalcificadas. No obstante, cuando los márgenes de las restauraciones se volvían visibles, eran reparados en los controles, lo cual contribuía a la satisfacción de la paciente.
Consideramos de gran importancia, que en la progresiva resolución exitosa de este caso, y que aún requerirá nuevas etapas, la influencia de factores sociales y funcionales positivos de la paciente y su entorno fueron fundamentales (familia funcional, estilos de vida saludable, acceso a la salud, etc). Estos aspectos son contemplados en el nuevo paradigma biopsicosocial de la salud bucal, como factores moderadores y determinantes conductoras, a los efectos de promover y mantener la salud bucal. 1-2
Conclusiones
La amelogénesis imperfecta, constituye un grupo de trastornos heterogéneos, tanto clínicos como genéticos, de naturaleza hereditaria. Las múltiples alteraciones que produce alcanzan la salud bucal, la emocional y en definitiva a la calidad de vida, lo que hace necesaria la atención integral del paciente. El abordaje interdisciplinario, con la colaboración temprana, estrecha y mantenida en el tiempo de los profesionales de la salud, el paciente y su entorno familiar permitió en la paciente presentada optimizar los resultados del complejo tratamiento. El plan de tratamiento se basó, como recomienda la literatura, en factores como edad, tipo de defectos y necesidades individuales. Se priorizó restablecer la estética y controlar la sensibilidad a fin de generar bienestar general y emocional reforzando la autoestima del paciente, que se hizo evidente por la aceptación del tratamiento con una sonrisa alentadora. El éxito a largo plazo dependerá de la adherencia a los controles periódicos estrictos, para lo que se requerirá la motivación constante de la paciente y su entorno familiar, lo cual se viene cumpliendo satisfactoriamente en el caso clínico presentado en este artículo.
Referencias bibliográficas
-
Lee JY, Watt RG, Williams DM, Giannobile WV. A New Definition for Oral Health: Implication for Clinical Practice, Policy and Research. J Dent Res. 2017; 96: 125-127.
-
FDI´s definition of oral health, October 2016. Recuperado de: http://www.fdiworlddental.org/oral-health/vision-2020/fdis-definition-of-oral-health
-
Poulter JA , Murillo G, Brookes SJ, Smith CE, Parry DA, Silva S, Kirkham J, Inglehearn CF, Mighell AJ. Deletion of ameloblastin exon 6 is associated with amelogenesis imperfecta. Hum Mol Genet. 2014; 23: 5317-24.
-
Witkop CJ Jr. Amelogenesis imperfecta, dentinogenesis imperfect and dentin dysplasia revisited: Problems in classification. J Oral Pathol. 1988; 17: 547-53.
-
Murillo G, Silva S, Mata M, Esquivel MJ. Amelogénesis Imperfecta. Probabilidad genética de expresión en futuras generaciones de familias costarricenses. Odovtos-Int. J. Dental S.C. 2014, 16: 71-86.
-
Prasad MK, Laouina S, El Aloussi M, Dollfus H, Bloch-Zupan A. Amelogenesis Imperfecta: I Family, 2 Phenotypes and 2 Mutated genes. J Dent Res. 2016; 95: 1457-63.
-
Smith C, Murillo G, Brookes SJ, Poulter JA, Silva S, Kirkham J, Inglehearn CF, Mighell AJ. Deletion of amelotin exons 3–6 is associated with amelogenesis imperfect. Hum Mol Genet. 2016; 25: 3578–87.
-
Koruyucu M, Bayram M, Tuna EB, Gencay K, Seymen F. Clinical Findings and long-term managements of patients with amelogenesis imperfecta. Eur J Dent. 2014; 8: 546-52.
-
Ortega Urzua O, Rodríguez P, Martínez Morales B. Análisis genético-clínico de una familia afectada con una malformación de esmalte dental. Rev Médica Chilena 2015; 133: 1331-40.
-
Neville BW, Damm DD, Allen CM, Chi AC. Abnormalities of teeth. En: Oral and Maxillofacial Pathology. 4th ed. St. Louis, MO: Elsevier Saunders; 2016: cap 2.
-
Gadhia K., McDonald S., Arkutu N., Malik K. Amelogénesis imperfecta: an introduction. Br Dent J. 2012; 212: 377-9.
-
Poulsen S, Gjørup H, Haubek D, Haukali G, Hintze H, Løvschall H, Errboe M. Amelogenesis imperfecta-a systematic literature review of associated dental and oro-facial abnormalities and tact on patients. Acta Odontol Scand. 2008; 66: 193-9.
-
Coffield K, Phillips C, Brady M, Roberts MW, Strauss RP, Wright JT. The psychosocial impact of development dental defects in people with hereditary amelogenesis imperfecta. J Am Dent Assoc. 2005; 136: 620–30.
-
Murillo G, Morales F, Gamboa LC, Meza AM, López AC. Impacto emocional y en calidad de vida de individuos afectados por amelogénesis imperfecta. Odovtos-Int. J. Dent. Sc. 2015; 17: 73-85.
-
Parekh S, Almehateb M, Cunningham S. How do children with amelogénesis imperfect feel about their teeth? Int. J. Paediatr. Dent. 2014; 24: 326–335.
-
Montero J, Bravo M, Albaladejo A, Hernández LA, Rosel EM. Validation the Oral Impact Profile (OHIP-14sp for adults in Spain. Med Oral Patol Oral Cir Bucal. 2009 1; 14: 44-50.
-
Lata S, Varghese NO, Varughese JM. Remineralization potential of fl uoride and amorphous calcium phosphate-casein phosphor peptide on enamel lesions: An in vitro comparative evaluation. J Conserv Dent. 2010; 13: 42-6.
-
Huq NL, Myroforidis H, Cross KJ, Stanton DP, Veith PD, Ward BR, Reynolds EC. The Interactions of CPP–ACP with Saliva. Int J Mol Sci. 2016 9; 17: 915.
-
Azarpazhooh A, Limeback H. Clinical efficacy of casein derivatives: A systematic review of the literature. J Am Dent Assoc. 2008; 139: 915-24.
-
Reynolds EC, Cai F, Cochrane NJ, Shen P, Walker GD, Morgan MV. Fluoride and casein phosphopeptide-amorphous calciumphosphate. J Dent Res. 2008; 87: 344-8
-
Markovic D, Petrovic B, Peric T. Case series: Clinical findings and oral rehabilitation of patients with amelogenesis imperfecta. Eur Arch Paediatr Dent. 2010; 11: 201-8.
Recibido: 05/01/18
Aceptado: 02/05/18
Correspondencia: María del Carmen López Jordi
Fuente: https://www.revistaodontopediatria.org/ediciones/2019/1/art-7/
Amelogénesis imperfecta
Es un trastorno del desarrollo dental. Causa que el esmalte del diente sea más delgado y se forme de manera anormal. El esmalte es la capa externa de los dientes.
Causas
La amelogénesis imperfecta se transmite de padres a hijos como un rasgo dominante. Eso significa que usted solo necesita recibir el gen anormal de uno de los padres para tener la enfermedad
Síntomas
El esmalte de los dientes es suave y delgado. Los dientes se ven amarillos y se dañan fácilmente. Tanto los dientes de leche como los permanentes pueden resultar afectados.
Pruebas y exámenes
Un odontólogo puede identificar y diagnosticar la afección.
Tratamiento
El tratamiento depende de la gravedad del problema. Las coronas dentales completas pueden ser necesarias para mejorar la apariencia de los dientes y protegerlos de más daño. Consumir una alimentación que sea baja en azúcar y practicar una muy buena higiene oral puede reducir la posibilidad de formación de caries.
Expectativas (pronóstico)
El tratamiento es generalmente efectivo para proteger los dientes.
Posibles complicaciones
El esmalte se daña fácilmente, lo cual afecta la apariencia de los dientes, especialmente si se deja sin tratamiento.
Cuándo contactar a un profesional médico
Consulte con el odontólogo si tiene síntomas de esta afección.
Referencias
Neville BW, Damm DD, Allen CM, Chi AC. Abnormalities of teeth. In: Neville BW, Damm DD, Allen CM, Chi AC, eds. Oral and Maxillofacial Pathology. 4th ed. St Louis, MO: Elsevier; 2016:chap 2.
Tinanoff N. Development and developmental anomalies of the teeth. In: Kliegman RM, Stanton BF, St. Geme JW, Schor NF, eds. Nelson Textbook of Pediatrics. 20th ed. Philadelphia, PA: Elsevier; 2016:chap 307.
Ultima revisión 2/5/2018
Versión en inglés revisada por: Ilona Fotek, DMD, MS, Dental Healing Arts, Jupiter, FL. Review provided by VeriMed Healthcare Network. Also reviewed by David Zieve, MD, MHA, Medical Director, Brenda Conaway, Editorial Director, and the A.D.A.M. Editorial team.
Traducción y localización realizada por: DrTango, Inc.
Fuente: https://medlineplus.gov/spanish/ency/article/001578.htm
Amelogénesis imperfecta en una familia
Revista Cubana de Estomatología
versión impresa ISSN 0034-7507versión On-line ISSN 1561-297X
Rev Cubana Estomatol vol.55 no.2 Ciudad de La Habana abr.-jun. 2018
PRESENTACIÓN DE CASOS
Amelogenesis imperfecta in a family
Paula Hurtado-Villa,I Fabián Tobar-Tosse,I Julio Osorio,II Freddy MorenoI
I Facultad de Ciencias de la Salud. Pontificia Universidad Javeriana Cali. Colombia.
II Institución Universitaria Colegios de Colombia.
RESUMEN
Introducción: la amelogénesis imperfecta consiste en un grupo de desórdenes hereditarios que afectan el desarrollo del esmalte dental, de tal forma que se ve comprometida la apariencia clínica de todos o casi todos los dientes, tanto temporales como permanentes.
Objetivo: informar las características y condiciones clínicas de la dentición de tres individuos de una misma familia con diagnóstico presuntivo de amelogénesis imperfecta.
Presentación de casos: se realizó examen intrabucal a tres individuos con rango de consanguinidad de primer grado (madre y dos hijos) quienes presentaban alterado estructuralmente el esmalte de los dientes. De acuerdo con las características clínicas dentales y el método de Witkop, los individuos fueron diagnosticados de forma presuntiva con amelogénesis imperfecta hipomadura tipo II (madre), caracterizada por hipomaduración del esmalte y fragmentación por desgaste en los bordes incisales; amelogénesis imperfecta hipoplásica tipo I (hijo mayor), con amplias zonas de dentina expuesta opaca y con manchas pardas generalizadas; y amelogénesis imperfecta hipomadura tipo II (hijo menor), con predominio de lesiones en forma de copo de nieve o motas de algodón.
Conclusiones: el diagnóstico clínico de la amelogénesis imperfecta basado en métodos fenotípicos resulta impreciso debido a la imposibilidad de establecer el origen de las alteraciones macroestructurales del esmalte. Sin embargo, de acuerdo con la descripción de los tres casos, son las afecciones en la cantidad y calidad del esmalte las que permiten realizar un diagnóstico clínico presuntivo, que guía la implementación de un tratamiento odontológico direccionado a la solución del compromiso estético y a la prevención del compromiso del órgano dentino-pulpar. En esta presentación de casos, la manifestación fenotípica de la enfermedad pasó de la madre a ambos hijos, siendo la amelogénesis imperfecta hipomadura dominante en el hijo menor.
Palabras clave: amelogénesis imperfecta; esmalte dental; amelogénesis.
ABSTRACT
Introduction: amelogenesis imperfecta consists of a group of hereditary disorders that affect the development of the dental enamel in such a way that the clinical appearance of all or almost all primary and permanent teeth is compromised.
Objective: report the clinical characteristics and conditions of the dentition of three individuals from the same family with a presumptive diagnosis of amelogenesis imperfecta.
Case presentation: intraoral examination was performed of three first-degree relatives (mother and two children) with structurally altered tooth enamel. Based on their clinical dental characteristics and the results of the Witkop method, the individuals were presumptively diagnosed with hypomaturation amelogenesis imperfecta type II (mother), characterized by enamel hypomaturation and fragmentation by wear on the incisal edges; hypoplastic amelogenesis imperfecta type I (elder son), with large areas of opaque exposed dentin and generalized brown spots; and hypomaturation amelogenesis imperfecta type II (younger son), with a predominance of lesions in the shape of snowflakes or cotton wads.
Conclusions: clinical diagnosis of amelogenesis imperfecta based on phenotypic methods is imprecise, since it is not possible to establish the origin of the macrostructural alterations of the enamel. However, according to the description of the three cases, quantitative and qualitative damage to the enamel makes it possible to establish a presumptive clinical diagnosis which will guide the implementation of a dental treatment aimed at resolving the aesthetic commitment and preventing involvement of the dentine-pulp complex. In this case presentation, the phenotypic manifestation of the disease passed from the mother to both children, and hypomaturation amelogenesis imperfecta was dominant in the younger son.
Keywords: amelogenesis imperfecta; tooth enamel; amelogenesis.
INTRODUCCIÓN
La amelogénesis imperfecta (AI) constituye un grupo desórdenes hereditarios que afectan el desarrollo del esmalte, lo que altera su cantidad (macroestructura anatómica) y calidad (microestructura histológica),1 afectando el aspecto clínico de todos o casi todos los dientes, tanto deciduos como permanentes.2 Fue descrita inicialmente por Weinmann en 1945 (citado por Belmont y López en 1998)3 como una anomalía de desarrollo del esmalte dental de origen ectodérmico, debido a que los tejidos dentales de origen mesodérmico (dentina, cemento y pulpa) se encuentran normales. En la actualidad, se sabe que el origen de esta condición patológica se produce por una alteración a nivel genético durante la amelogénesis.4
La AI ha sido descrita como una alteración dominante o recesiva, tanto autosómica como ligada al cromosoma X, por lo que es considerada como un desorden genético heterogéneo en el que están envueltas diferentes mutaciones en uno o diferentes genes.5-7 En los seres humanos, defectos en el gen AMELX causan AI ligada al cromosoma X, mientras que mutaciones en el gen ENAM provocan AI autosómica dominante.8,9 Asimismo, se han identificado otros genes que pueden participar activamente en el desarrollo de este síndrome, entre los que se encuentra DLX3, FAM83H, MMP-20, KLK4 y WDR72.10 Si bien en un inicio los estudios se centraron en la descripción de la alteración histológica del esmalte, una vez determinado su origen genético surgió el interés por la identificación de los genes implicados en procura de definir la etiología. Hoy en día la investigación se ha redireccionado a la asociación de la AI con otras alteraciones en diferentes regiones del cuerpo, sin embargo, al revisar la literatura disponible (en su mayoría presentaciones de casos), se deduce que la AI es una condición de rara aparición, siendo la proporción de 1:700 a 1:16.000 casos,11-13 con alguna prevalencia hacia poblaciones de origen caucasoide.14,15
Por tanto, el objetivo de este artículo es informar las características y condiciones clínicas de la dentición de tres individuos de una misma familia con diagnóstico presuntivo de amelogénesis imperfecta.
PRESENTACIÓN DE CASOS
Se estudiaron tres pacientes con rango de consanguinidad en primer grado, residentes de un sector de estrato socio-económico bajo de Cali (Colombia) y que asistieron a las clínicas odontológicas de una institución universitaria de Cali para examen clínico y tratamiento odontológico. Para la participación en el estudio se contó con el aval de un Comité de Ética en Investigación y con la firma del consentimiento informado por parte de los individuos. El motivo de la consulta fue la preocupación de la madre por sus dientes amarillos y la presencia de esta misma característica en sus hijos. Reporta que continuamente se les realizaron restauraciones dentales que en pocos días se desalojaban de su cavidad dental. El diagnóstico clínico de la AI se realizó con ayuda de varios especialistas en odontopediatría, teniendo en cuenta los posibles sesgos entre hipomaduración del esmalte y pérdida del esmalte, debido a la edad de los pacientes. Para la clasificación de la AI se empleó el método de Witkop de 1998.14
CASO 1
Paciente femenino de 35 años de edad, que corresponde a la madre y que al examen clínico presenta zonas de dentina expuesta en los tercios incisales de los dientes anteriores inferiores asociadas a hipomaduración del esmalte y fragmentación por desgaste en los bordes incisales. En los dientes posteriores se observa exposición de la dentina asociada a hipomaduración y fragmentación del esmalte en la zona oclusal. En los dientes anteriores, el esmalte se observa blanco y con pérdida de brillo por hipomaduración; mientras que en los posteriores se observa amarillo, con pérdida de brillo y de aspecto rugoso por descalcificación. Se presumen que las fragmentaciones ocurren a nivel de la unión amelodentinaria (falla adhesiva), bien porque el esmalte no se formó; o si lo hizo, no desarrolló un patrón de unión festoneado adecuado, por lo que posteriormente se fragmentó durante la función masticatoria exponiendo la dentina al desgaste. Diagnóstico presuntivo: amelogénesis imperfecta del tipo hipomadura Tipo II (Fig. 1).
CASO 2
Paciente masculino de 13 años de edad, que corresponde al hijo mayor y que al examen clínico presenta el esmalte de todos los dientes superiores e inferiores permanentes afectados de alguna forma. Existe ausencia de desarrollo del esmalte con amplias zonas de dentina expuesta, esta última opaca y con manchas pardas generalizadas. En algunos casos en donde el esmalte persiste se observa blanco y brillante, mientras que otras veces se observa opaco, rugoso y discontinuo por descalcificación. Se presumen que la ausencia, fragmentación y perforaciones del esmalte ocurren a nivel de la unión amelodentinaria (falla adhesiva), sin involucrar a la dentina. Es posible identificar lesiones de esmalte hipomaduro e hipomineralizado. Se pueden observar diastemas dentales asociados a fragmentación del esmalte interproximal por disminución de su resistencia en la distribución de las fuerzas transversales durante la función de masticación y disminución del volumen de la corona por excesivo desgaste. Diagnóstico presuntivo: amelogénesis imperfecta del tipo hipoplásica Tipo I (Fig. 2).
CASO 3
Paciente masculino de 9 años de edad, que corresponde al hijo menor y que al examen clínico presenta el esmalte de todos los dientes permanentes afectados de alguna forma, asociados a lesiones de hipocalcificación. Predominan las lesiones en forma de copo de nieve o motas de algodón, sobre todo en las superficies vestibulares, con perforaciones asociadas a hipoplasias puntuales que traslucen la dentina subyacente de color amarillo pardo. Diagnóstico presuntivo: amelogénesis imperfecta del tipo hipomadura Tipo II (Fig. 3).
DISCUSIÓN
La descripción de las lesiones aquí presentadas resulta compatible con descripciones encontradas en la literatura. El análisis clínico permitió identificar lesiones hipocalcificantes evidentes en los casos 1 y 3 (esmalte blando, rugoso y de color pardo); aunque en el primero, dada la edad de la paciente, la ausencia del esmalte en algunas regiones se puede asociar a fragmentación y desgaste durante la función masticatoria y el cepillado; y una serie de lesiones hipoplásicas del esmalte en los tres casos en donde el esmalte evidencia alteración en el espesor y textura irregular con presencia de hoyos. Sin embargo, la dureza y transparencia son normales. Lesiones compatibles con esmalte hipomaduro se relacionan con manchas blancas opacas.1,16 Algunos reportes concluyen que los diferentes fenotipos de AI pueden combinarse y dificultar el diagnóstico, para lo cual se requiere definir los patrones de herencia.6
PATRONES MORFOGENÉTICOS
Genéticamente, la AI ligada al cromosoma X se transmite de forma autosómica dominante o recesiva.1,9 Cuando se encuentran ligadas al cromosoma X se asocian a mutaciones en el gen AMEL, las cuales afectan la cantidad de esmalte (hipoplasia) y/o defectos en su mineralización (hipomaduración).6,17 De esta forma, el diagnóstico que corresponde a AI hipoplásica o combinada, es transmitida por la madre portadora de AI en un 50 % a sus hijos de ambos géneros, siendo mayor el compromiso del esmalte afectado en los individuos masculinos.16 En este caso, la manifestación fenotípica de la enfermedad pasó de la madre a ambos hijos, siendo la AI hipomadura dominante en el individuo de 9 años de edad.
En la AI transmitida de forma autosómica dominante, la alteración ocurre en el gen ENAM del cromosoma cuatro, afectando el proceso de mineralización del esmalte en las primeras etapas de la amelogénesis. Los diagnósticos que resultan puede ser AI hipoplásica generalizada en la que el aspecto del esmalte es liso, delgado, con bandas horizontales y hoyos en la superficie, similar al caso 2 de AI hipoplásica localizada, caracterizada por bandas horizontales de hoyos que abarcan parte del diente afectado.6,9 La AI autosómica dominante se corresponde clínicamente con las formas hipoplásica, hipoplásica con hipocalcificación o hipomadurativa, la cual afecta a uno o más individuos en cada generación de una misma familia con compromiso del esmalte variable.6 En el caso de la AI autosómica recesiva, la alteración ocurre en el gen de la MMP-20 y en el gen de la KLK-4, los cuales participan en las fases de secreción y maduración de la amelogénesis.16 Las mutaciones en estas proteasas se asocian con AI hipomadura e hipomadura pigmentada, caracterizadas por un esmalte de espesor normal, desmineralizado y con pigmentaciones de color amarillo-café, similar a los casos 1 y 3 de esta presentación.
En la actualidad, el avance en el conocimiento y la comprensión de la fisiopatología de la AI se ve limitado por la diversidad genética y la baja prevalencia. Además, debido al costo del análisis genético como método diagnóstico, resulta imposible aplicarlo de forma rutinaria. Sin embargo, es posible deducir en la revisión de la literatura, que se están estableciendo correlaciones fenotipo-genotipo a través del estudio clínico y el análisis genético molecular de familias afectadas.1
SEPARACIÓN DEL ESMALTE A NIVEL DE LA UNIÓN AMELODENTINARIA
Durante la odontogénesis, la unión amelodentinaria (UAD) se comporta como la plataforma de inicio de la amelogénesis y de la dentinogénesis configurando una interfase festoneada entre el esmalte y la dentina, mucho más imbricada en la región de las cúspides.18,19 Esta UAD disipa las fuerzas generadas durante la masticación, optimizando el comportamiento microestructural de ambos tejidos.20 Por tanto, si hay una alteración genética en la síntesis de los componentes de la matriz extracelular desde el inicio de la amelogénesis, la UAD no quedará conformada de forma adecuada, lo que afecta el patrón festoneado y ocasiona que ante la función masticatoria se venza el escaso límite de resistencia y se fragmente el esmalte en la unión con la dentina. Esto asociado a que la microdureza del esmalte se ve afectada, mucho más en el fenotipo hipocalcificado que en el hipomaduro,21 tal como se puede observar en las figuras 1 y 2.
Dicha fragmentación resulta evidente en las superficies vestibulares y linguales donde la UAD morfológicamente es menos festoneada en los bordes incisales y vértices cuspídeos por la atrición y en la región cervical ante el cambio dirección y menor longitud de los prismas de esmalte por abfracción. En contraste, en los pacientes con fenotipo hipomaduro que mantienen el esmalte afectado poroso y adherido a la dentina, como se observa en la figura 3, la alteración de los prismas ocurre en la superficie, por lo que se considera que la AI ocurrió en la fase de maduración, razón por la cual los defectos estructurales ocasionados por la constitución de un esmalte aprismático se asocian con la disminución de actividad de los ameloblastos y el deterioro del proceso de Tomes durante el ensamblaje final del prisma.21
CONSIDERACIONES CLÍNICAS
Si bien es cierto que las alteraciones del esmalte en la AI no implican un riesgo vital de los pacientes, estas pueden impactar su calidad de vida debido al compromiso estético. Por lo general, la descripción clínica de un paciente portador de AI incluye una estética dental deficiente, sensibilidad térmica alta, desgaste dentario extenso, caries secundaria, decoloración dentaria, maloclusiones y compromiso periodontal; además de alteraciones psicosociales asociadas por fenotipos con mayor compromiso estético. De allí la importancia del manejo integral y multidisciplinario de los pacientes, del diagnóstico temprano y de un tratamiento restaurador y rehabilitador adecuado. Sin embargo, es la atención preventiva —teniendo en cuenta el riesgo de caries, la fragmentación posteruptiva del esmalte y exposición de la dentina, la presencia de sensibilidad dental, la etiología de la enfermedad y la gravedad (color y presencia, tamaño y profundidad de las fracturas en el esmalte) y extensión (número de dientes afectados)—, la que permitirá adaptar el plan de tratamiento a cada caso en particular, con el objetivo de mejorar la estética (operatoria dental), reducir la sensibilidad (aplicación tópica de flúor), corregir o mantener la dimensión vertical (rehabilitación bucal u ortodoncia) y restablecer las diferentes funciones del sistema estomatognático, incluida la masticación.22-25
Se concluye que la AI es un desorden hereditario que afecta el desarrollo del esmalte de tal forma que altera la cantidad y calidad macro y microestructural. Las alteraciones permiten realizar un diagnóstico presuntivo que guiará hacia la implementación de un tratamiento odontológico que solucione de mejor manera el compromiso estético y el compromiso del órgano dentino-pulpar. Las clasificaciones actuales, incluida la de Witkop, se basan fundamentalmente en el fenotipo,14,15 lo genera dificultades al momento del diagnóstico, dado que las alteraciones clínicas del esmalte como la hipoplasia, hipomaduración e hipocalcificación, se pueden presentar de forma específica o generalizada en los diferentes grupos de dientes, desarrollando una amplia variabilidad de compromisos morfofuncionales que dificultan la clasificación y, por ende, el pronóstico del plan de tratamiento odontológico. Es por ello que se deben emplear métodos diagnósticos basados en el genotipo para confirmar el origen genético de la AI, lo que permitiría valorar fehacientemente la naturaleza de las lesiones del esmalte y lograr un pronóstico favorable, a partir del manejo integral de los pacientes desde la asesoría genética hasta los tratamientos odontológicos oportunos y acertados.
Agradecimientos
Esta presentación de casos fue desarrollada dentro del proyecto de investigación "Análisis genético, clínico y molecular de una familia afectada con amelogénesis imperfecta" financiado por la Convocatoria Interna 2013-2014 de la Pontificia Universidad Javeriana Cali.
Conflicto de intereses
No existe conflicto de intereses.
REFERENCIAS BIBLIOGRÁFICAS
1. Prasad MK, Laouina S, El Alloussi M, Dollfus H, Bloch-Zupan A. Amelogenesis Imperfecta: 1 Family, 2 Phenotypes, and 2 Mutated Genes. J Dent Res. 2016;95(13):1457-63.
2. Oliveira FV, Gurgel CV, Kobayashi TY, Dionísio TJ, Neves LT, Santos CF, et al. Amelogenesis imperfecta and screening of mutation in amelogenin gene. Case Rep Dent. 2014;2014:319680.
3. Belmont C, López PM. Amelogénesis imperfecta del tipo hipomaduración-hipoplasia con taurodontismo. Rev División de Estudios de Posgrado e Investigación. 1998;2(8):18-22.
4. Sabandal MM, Schäfer E. Amelogenesis imperfecta: Review of diagnostic findings and treatment concepts. Odontology. 2016;104(3):245-56.
5. Wright JT, Hart PS, Aldred MJ, Seow K, Crawford PJM, Hong SP, et al. Relationship of phenotype and genotype in X-Linked amelogenesis imperfecta. Connect Tissue Res. 2003;44(1):72-8.
6. Wright JT. The molecular etiologies and associated phenotypes of amelogenesis imperfecta. Am J Med Genet A. 2006;140(23):2547-55.
7. Crawford PJ, Aldred M, Bloch-Zupan A. Amelogenesis imperfecta. Orphanet J Rare Dis. 2007;2(17):1-11.
8. Kim YJ, Kim YJ, Kang J, Shin TJ, Hyun HK, Lee SH, et al. A novel AMELX mutation causes hypoplastic amelogenesis imperfecta. Arch Oral Biol. 2017;76:61-5.
9. Stephanopoulos G, Garefalaki ME, Lyroudia K. Genes and related proteins involved in amelogenesis imperfecta. J Dent Res. 2005;84:1117-26.
10. Urzúa B, Ortega-Pinto A, Morales-Bozo I, Rojas-Alcayaga G, Cifuentes V. Defining a new candidate gene for amelogenesis imperfecta: From molecular genetics to biochemistry. Biochem Genet. 2011;49(1-2):104-21.
11. Mehta DN, Shah J, Thakkar B. Amelogenesis imperfecta: Four case reports. J Nat Sci Biol Med. 2013;4(2):462-5.
12. Gopinath V, Al-Salihi K, Yean Yean C, Chan M, Li A, Ravichandran M. Amelogenesis imperfecta: Enamel ultrastructure and molecular studies. J Clin Pediatr Dent. 2004;28(4):319-22.
13. Leme MC, Peres SR. The genetics of amelogenesis imperfecta: A review of the literature. J App Oral Sci. 2005;13(3):212-7.
14. Witkop CJ. Amelogenesis imperfecta, dentinogenesis imperfecta and dentin dysplasia revisited: Problems in classifications. J Oral Pathol. 1998;17:547-53.
15. Hurtado PM, Tobar-Tosse F, Osorio J, Orozco L, Moreno F. Amelogénesis imperfecta: Revisión de la literatura. Rev Estomatol. 2015;23(1):32-41.
16. Harryparsad A, Rahman L, Bunn BK. Amelogenesis imperfecta: a diagnostic and pathological review with case illustration. SADJ. 2013;68(9):404-7.
17. Büchi D, Fehmer V, Sailer I, Wolleb K, Jung R. Minimally invasive rehabilitation of a patient with amelogenesis imperfecta. Int J Esthet Dent. 2014;9(2):134-45.
18. Sa Y, Liang S, Ma X, Lu S, Wang Z, Jiang T, et al. Compositional, structural and mechanical comparisons of normal enamel and hypomaturation enamel. Acta Biomater. 2014;10(12):5169-77.
19. Radlanski RJ, Renz H. Insular dentin formation pattern in human odontogenesis in relation to the scalloped dentino-enamel junction. Ann Anat. 2007;189:243-50.
20. Moreno F, Ortiz M, Mejía C. Métodos de separación y técnicas de observación microscópica de la unión amelodentinaria: revisión sistemática de la literatura. Univ Odontol. 2013;32(69):19-34.
21. Wright JT, Hall KI, Yamauch M. The enamel proteins in human amelogenesis imperfecta. Arch Oral Biol. 1997;42(2):149-59.
22. Marquezin MC, Zancopé BR, Pacheco LF, Gavião MB, Pascon FM. Aesthetic and functional rehabilitation of the primary dentition affected by amelogenesis imperfecta. Case Rep Dent. 2015;2015:790890.
23. Ergun G, Kaya BM, Egilmez F, Cekic-Nagas I. Functional and esthetic rehabilitation of a patient with amelogenesis imperfecta. J Can Dent Assoc. 2013;79:d38.
24. Pousette Lundgren G, Wickström A, Hasselblad T, Dahllöf G. Amelogenesis imperfecta and early restorative crown therapy: An interview study with adolescents and young adults on their experiences. PLoS One. 2016;11(6):e0156879
25. Moura CDVS, Pontes AS, Lopes TSP, Moura LFAD, Lima MDM. Functional and esthetic rehabilitation of a child with amelogenesis imperfecta: A case report. Gen Dent. 2017;65(3):18-20.
Recibido: 10 de mayo de 2017.
Aprobado: 3 de marzo de 2018.
Fuente: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0034-75072018000200007
Intervenciones para la restauración de la amelogénesis imperfecta en niños y adolescentes
La amelogénesis imperfecta (AI) es un trastorno que ocurre durante el desarrollo dentario, en el cual los dientes son cubiertos por un esmalte delgado que se forma de manera anormal. El esmalte se fractura y se daña fácilmente, lo cual afecta la apariencia de los dientes, especialmente si se no se trata. Se encontró que los resultados psicológicos negativos, al comprometer la apariencia y la función en los pacientes con AI, afectan el atractivo de la persona y reducen la interacción social. La prevención y restauración tempranas y apropiadas son esenciales para el tratamiento exitoso de la AI y para el bienestar psicológico del paciente.
El tratamiento utilizado depende de la gravedad del problema. A veces, se utilizan coronas para mejorar la apariencia de los dientes y protegerlos del daño.
Esta revisión realizada por el Grupo Cochrane de Salud Oral intentó comparar las tasas de éxito de diferentes materiales restaurativos y de las técnicas utilizadas para la restauración de los dientes frontales y posteriores afectados por AI. El objetivo de la revisión fue evaluar la satisfacción del paciente, en particular, el aspecto de los dientes, su sensibilidad y su función.
La búsqueda más reciente de los estudios existentes se realizó el 18 de abril de 2013. Se realizaron búsquedas de ensayos controlados aleatorios en los que las restauraciones de los dientes afectados por AI se compararon con respecto a la satisfacción del paciente y la función. Los niños y adolescentes menores de 18 años de edad, que presentaban AI y necesitaban restauración, fueron seleccionados para la inclusión de forma independiente de su nacionalidad. Esta revisión no encontró ensayos que cumplieran los criterios de inclusión.
Por lo tanto, las pruebas fiables actuales son insuficientes para decidir cuál de estos tratamientos es el más efectivo. Deben realizarse ensayos controlados aleatorios bien definidos que se centren en niños y adolescentes con diferentes tipos y gravedad del trastorno para determinar la mejor intervención para restaurar los dientes afectados por AI.
Conclusiones de los autores:
No se encontraron ensayos controlados aleatorios de los tratamientos restaurativos en niños y adolescentes con AI, y por lo tanto, no hay pruebas sobre cuál es la restauración más adecuada. Deben realizarse ensayos controlados aleatorios bien definidos que incluyan a niños y adolescentes y se centren en el tipo y gravedad del trastorno para determinar la mejor intervención para restaurar los dientes afectados por AI.
Leer el resumen completo…
Notas de traducción:
La traducción y edición de las revisiones Cochrane han sido realizadas bajo la responsabilidad del Centro Cochrane Iberoamericano, gracias a la suscripción efectuada por el Ministerio de Sanidad, Servicios Sociales e Igualdad del Gobierno español. Si detecta algún problema con la traducción, por favor, contacte con Infoglobal Suport, cochrane@infoglobal-suport.com.
También puede estar interesado en:

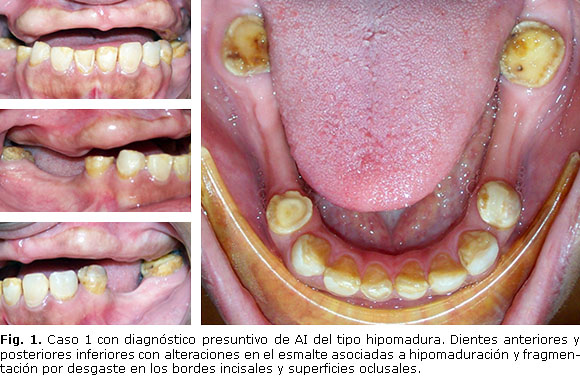{kind=link}
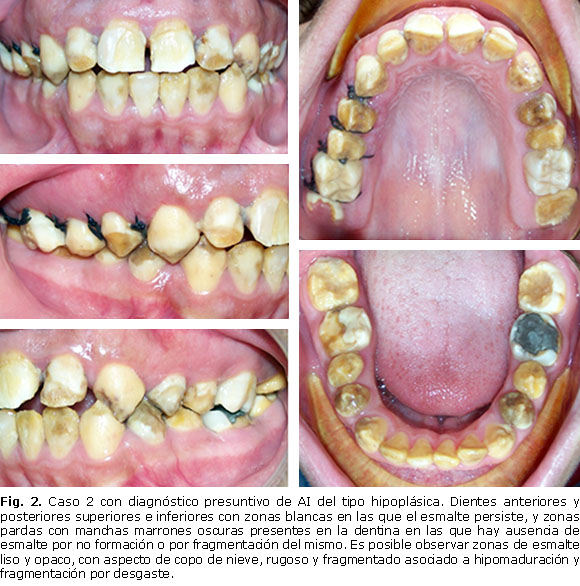{kind=link}
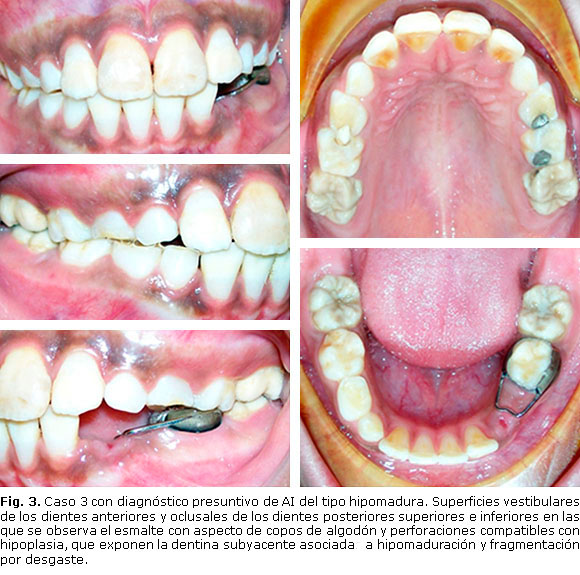{kind=link}